






















Summary
India has the second largest population in the world and ~60,000 children are born every day. Due to the high birth rate in the country, a large number of infants are born with congenital malformations, genetic conditions like inborn error of metabolic disorders (IEM). Many of these IEMs are treatable if diagnosed at an early age. Interestingly, there are a few conditions like congenital hypothyroidism (CH), congenital adrenal hyperplasia (CAH), Glucose 6 phosphate dehydrogenase (G6PD) deficiency that are treatable with a minimum of cost if identified early, which is the primary norm for new born screening (NBS). Hence, the presence of a screening program that screens newborn babies for common treatable conditions is essential to identify affected babies early. This shall aid in significant reduction of infant morbidity, mortality in the country with a significant reduction in national health burden. The present article describes the key points of a successful newborn screening program. It emphasizes the need for setting up a centralized screening program in India for the treatable genetic conditions: congenital hypothyroidism, congenital adrenal hyperplasia and glucose-6-phosphate dehydrogenase deficiency.
Conflict of Interest: None declared
Source of Support: Ms. Aadhira Nair is a junior research fellow on a research project funded by Gujarat State Biotechnology Mission (GSBTM), Govt of Gujarat, India.
Introduction
Newborn screening (NBS) is a simple and cost-effective strategy for the early identification of inborn error of metabolic disorders (IEM) in the neonates immediately after birth. The idea of NBS was first described by Guthrie in 1962 for screening of phenylketonuria using heel prick blood samples on filter paper.[1] Gradually, screening strategies have been developed for several other genetic conditions like congenital adrenal hyperplasia (CAH), congenital hypothyroidism (CH), glucose-6-phosphate dehydrogenase (G6PD deficiency), galactosemia and many others. The basic idea of carrying out NBS is to identify newborn with common treatable conditions prior to the manifestation of symptoms. Early treatment in these infants can prevent or minimize serious irreversible complications like mental retardation, metabolic complications and disabilities. This shall have a direct consequence on a significant reduction of infant morbidity and mortality. Additionally, in the presence of a successful NBS, the financial burden incurred on the families and the society will be far less as compared to that without NBS because these tests are cheap and the available treatment options have a good prognosis.
The key components for an effective NBS include: a) educating the participating families about the benefits of the screening program, b) timely sample collection, c) recruitment of trained professionals for performing appropriate tests, d) generation of reports and conveying the same to the patients, e) providing counselling when required, and f) monitoring the long-term outcome and cost-effectiveness of the screening program.
Here, we present an overview of NBS in India and emphasize its role specifically in context to three common affordable treatable genetic conditions namely congenital hypothyroidism (CH), congenital adrenal hyperplasia (CAH), and glucose-6-phosphate dehydrogenase deficiency (G6PD).
Need for NBS in India
NBS is a mandatory procedure in many developed countries like USA, UK, Australia, Sweden, Finland and Japan for screening as many as 54 different inherited conditions.[2] However, despite India having a high birth rate of ~60,000 children born every day, NBS is not a routine practice.[3] Moreover, data from previously reported studies show a high incidence of conditions like CH (1:1,103), CAH (1:5,762), and G6PD (1:66-1:1,000).[4-6] A high rate of consanguinity and endogamy in many communities in India, as well as a lack of awareness and lack of adequate diagnostic facilities in many parts of the country, are likely to be the major contributors to the high incidence of IEM. Nonetheless, this emphasizes the need for implementing a national-level centralized NBS program. The major hurdles of implementation of an effective NBS in India are: a) unavailability of accurate epidemiology data of common treatable conditions for the Indian population in different regions of the country, b) lack of awareness of treatable genetic conditions in the medical community, c) dearth of necessary infrastructure and trained manpower for the program, and d) absence of a national policy for NBS.
Status of NBS in India
Dr Meena Desai was the first one to initiate NBS for CH in the country in the early eighties in Mumbai[7]followed by Dr Appaji Rao and colleagues who carried out the newborn metabolic screening program for amino acid disorders in Bangalore in 1988.[8] Likewise, Devi R and her group in 1994 screened 5140 neonates for G6PD deficiency and reported 7.8% cases to show G6PD deficiency.[9] Subsequently, several newborn screening studies were taken up in different parts of the country on a small scale. The first mass screening program was carried out in the union territory of Chandigarh in 2007. Newborns of 24-48hr of age were tested for three conditions namely CH, CAH, and G6PD[10]. Thereafter, ICMR, through a Task Force on Inborn Metabolic Disorders, had set up a screening program that lasted from 2007 to 2012. In this study, a total of 207,561 newborns were screened for CH and CAH from 5 centres in India, Delhi, Mumbai, Chennai, Hyderabad, and Kolkata. They observed that the incidence of CH varied from 1 in 727 in Chennai to 1 in 1,528 in Mumbai. Likewise, for CAH the incidence varied from 1 in 2,036 in Chennai to 1 in 9,983 in Mumbai.[4]
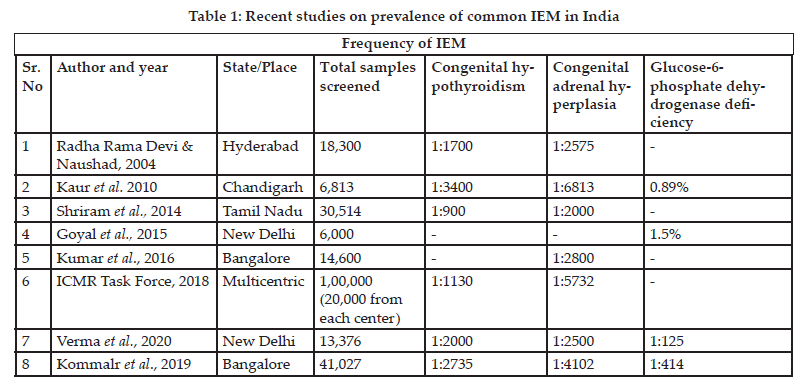 Goa became the first state in India to introduce mandatory NBS in 2008. The study data is available at dhsgoa.gov.in; however, the follow-up data is unavailable. In 2011-2012, the Gujarat government also initiated NBS in 7 medical colleges. In this program, only infants with a high suspicion index were screened that does not reflect the true incidences. Another pilot study was conducted in a rural district in Andhra Pradesh to screen 5000 newborns for metabolic disorders.[11] Thus, there have been several attempts to implement a successful screening program in different parts of the country. Table 1 gives an overview of the recent studies carried out across the country to determine the prevalence of common IEMs in India. The National Neonatology Forum recommends that NBS should be done in all babies for three disorders– CH, CAH, and G6PD deficiency. These conditions have been selected due to their high incidence in the Indian population.
Goa became the first state in India to introduce mandatory NBS in 2008. The study data is available at dhsgoa.gov.in; however, the follow-up data is unavailable. In 2011-2012, the Gujarat government also initiated NBS in 7 medical colleges. In this program, only infants with a high suspicion index were screened that does not reflect the true incidences. Another pilot study was conducted in a rural district in Andhra Pradesh to screen 5000 newborns for metabolic disorders.[11] Thus, there have been several attempts to implement a successful screening program in different parts of the country. Table 1 gives an overview of the recent studies carried out across the country to determine the prevalence of common IEMs in India. The National Neonatology Forum recommends that NBS should be done in all babies for three disorders– CH, CAH, and G6PD deficiency. These conditions have been selected due to their high incidence in the Indian population.
NBS for Congenital Hypothyroidism
Congenital hypothyroidism (CH) is a condition in which there is a deficiency of thyroid hormone at birth. The two major causes of CH are due to problems either in the thyroid gland development or thyroid hormone biosynthesis. CH can be classified into congenital permanent and transient forms depending on the underlying etiology. Permanent CH requires lifelong treatment as there is a persistent deficiency of the thyroid hormone. However, in the case of transient CH, there is temporary thyroid hormone deficiency at birth followed by normal production of thyroid hormone. Permanent CH can be due to defects in thyroid hormone synthesis due to absent or poorly developed thyroid gland, a genetic defect in thyroid hormone production or mother on antithyroid drugs during pregnancy, low iodine intake by mother during pregnancy or rarely due to resistance to thyroid hormone action. Transient CH is due to maternal or neonatal factors. Antithyroid medication, transplacental thyrotropin receptor-blocking antibodies, exposure to iodine deficiency or excess of iodine are examples of maternal factors. Similarly, neonatal factors include congenital liver haemangiomas, neonatal iodine deficiency, or excess of iodine. The exact cause of CH may remain unknown in many cases. The critical point in the case of management of CH is restoring thyroid function to normal and this does not necessarily require knowledge of the underlying cause.
The epidemiological data shows the variable incidence of CH in the country. In the early 1980s, NBS was carried out for CH at BJ Wadia Hospital, Mumbai using cord blood TSH and postnatal DBS T4. They reported a prevalence of 1:2481 and 1:2804 respectively.[12,13] Subsequently, NBS was undertaken for CH in different states of India by independent groups. In a study carried out at Sir Ganga Ram hospital, New Delhi from January 2008 to December 2017, a total of 13,376 newborns were screened for CH, CAH, and G6PD. They reported an incidence of 1:2000 for CH. In this study, they observed 100% sensitivity and 99.9% specificity with a false positive reporting rate of <0.015% for CH.[14] This study, however, focussed on the North Indian population. A similar new-born screening program was carried out in Bangalore, South India for a period of 3 years (January 2016-December 2018). They screened 41,027 babies and reported an incidence of 1:2,735 live births for CH.[15] A total of 15 babies were confirmed to have CH. Of note, all the babies were asymptomatic at birth except for jaundice in a few cases. The first multicentric study carried out by ICMR task force in more than one lakh neonates has identified the incidence of CH to be in 1:1130.[4] These observations emphasize the importance of a screening program for the early detection of CH-affected babies in the country.
Initial Signs and Symptoms of CH
Generally, newborn infants with CH remain undiagnosed at birth as the initial clinical features are often subtle. This is because there is the passage of maternal thyroid hormone across the placenta. After birth, the symptoms gradually develop. These include hoarse cries, constipation, prolonged jaundice, lethargy, delayed speech, delayed growth and feeding difficulties. A few infants with CH may have a palpable goitre. The noticeable features of an infant with CH are a puffy face, flat nasal bridge, macroglossia, and protuberant abdomen.
Sample and Screening Tests for CH screening
A cord blood or postnatal sample may be used when screening for CH. However, the critical point to note is the neonatal surge of TSH and T4 which is generally 30 minutes after birth in case of TSH and may persist for 48h to 72hr.[16] If the screen sample is taken during the surge, there is a high possibility of a false-positive result. Hence, it is recommended that either cord blood or postnatal sample after 72 hr should be used for screening for CH.
There are two main screening strategies for CH: primary T4 testing or primary TSH testing. Primary T4 testing has a high rate of false-positive results[17] as compared to the primary TSH testing which is more sensitive and specific for the diagnosis of CH.[18] However, primary TSH testing may miss infants with the delayed rise of TSH mostly seen in cases of preterm babies due to immaturity of the hypothalamic-pituitary-thyroid axis.[12] Overall, primary TSH-based CH screening is more practical and cost-effective and hence is recommended for NBS for CH.
NBS for Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia (CAH) is a group of genetic disorders caused due to mutation in genes encoding enzymes involved in cortisol production. The two forms of CAH: severe (classic) and mild (non-classic) have been described. The underlying cause of CAH in 95% of the cases is a deficiency of enzyme 21-hydroxylase (21-OH) encoded by the CYP21A2 gene. There is a reduced glucocorticoid and mineralocorticoid synthesis and elevated precursors, most notably 17-hydroxyprogesterone (17-OHP) in case of defective 21-OH hydroxylation. In the case of classic CAH, there can be two forms, salt-wasting and simple virilizing depending on the severity of aldosterone deficiency. In the case of salt-wasting phenotype, there is a complete absence of 21-OH activity. Hence, an affected newborn presents with a life-threatening adrenal crisis in the first 2 weeks of life. However, in the case of patients with simple virilizing CAH, there is 1-2% of 21-OH activity and minimal aldosterone production that prevents a neonatal crisis. Nonetheless, in both the cases, there is a marked elevation of the hormone known as 17-Hydroxy progesterone (17-OHP) and this can be measured by various techniques known as ELISA, RIA, FIA, CLIA or LC/MS.
Neonatal screening for classic CAH due to 21-OH deficiency was first done in Alaska in 1977. NBS for classic CAH in the newborn period was recommended by the European Society for Pediatric Endocrinology Working Group in 2002.[19] In India, the burden of CAH is significant and it remains the most common cause of female ambiguity and primary adrenal failure in India.[20,21] However, the incidence or prevalence of CAH in India is unknown at present, it seems to be much higher in southern India (1:2600).[22] A multicentric study in India found 18 out of 104,066 newborns screened to have CAH, suggesting a collective incidence of 1 in 5762.[4] Recent studies from the south and north parts of the country found the CAH incidence to be 1 in 2800 and 1 in 6334 respectively.[10,23]
Sample Collection and Screening Tests for CAH
Cord blood is easier to collect but it is not a recommended sample for CAH screening as the steroid surge at birth results in significantly higher 17-OHP levels. This can lead to difficulty in result interpretation. Hence, capillary blood samples collected by heel prick on a filter paper are ideal for 17-OHP measurements for NBS. The sample collection time is also critical since the 17-OHP levels are influenced by a number of factors like sex, mode of delivery, seasonality, gestational age, and birth weight.[24-26] The recommended sample collection time for NBS should be at least 24 hr later after birth, but can be taken till day 7 of life using heel-prick.[27] Additionally, in the case of preterm babies, screening should be done at 2 weeks and 4 weeks of age or at discharge from NICU to reduce the risk of missing any CAH case.[28]
The traditional methods used for CAH screening are radioimmunoassay, enzyme-linked immunosorbent assay, or time-resolved Fluro immunoassay to measure the 17-OHP levels. Studies suggest that there may be high false-positive rates with the immunoassay due to cross-reactivity and physiological changes in the 17-OHP concentration during the first few days of life.[29] As a result, 2018 Endocrine Society Clinical Practice Guidelines recommend a second-tier screen by liquid chromatography-tandem mass spectrometry (LC/MS-MS).[28]
NBS for Glucose-6-phosphate Dehydrogenase Deficiency
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an X-linked recessive genetic disorder caused due to mutation in the G6PD gene. It is the most common erythrocyte enzymopathy seen in ~400 million people worldwide.[30] As the condition is X-linked, males are more likely to be affected than females. The exact incidence of G6PD is unknown at present, however, earlier studies have shown the incidence ranging from 2-27% in the Indian population.[31]
The G6PD enzyme is involved in the pentose phosphate pathway and catalyzes the conversion of nicotinamide dinucleotide phosphate to its reduced form. Its major role is protecting erythrocytes from oxidative damage. The common clinical manifestation due to G6PD deficiency in newborns is an increased risk and early onset of neonatal hyperbilirubinemia. Irritability, lethargy, poor feeding, vomiting, and fever in newborns with hyperbilirubinemia may indicate kernicterus, which can be fatal if not treated.[32] Infants with G6PD deficiency are twice as likely to develop neonatal jaundice than the general population.[33] In G6PD deficient erythrocytes, haemolysis may be triggered by infection, certain foods like fava beans as well as by some medications.[34] Thus, early detection and prevention of haemolytic episodes (by avoiding the triggers) is a potential strategy for the treatment of this condition.
NBS for G6PD Deficiency
In a study carried out at Sir Ganga Ram Hospital, New Delhi from January 2008 to December 2017, the incidence of G6PD was reported to be 1:87 in males and 1:250 females.[14] However, they also observed a high false-positive rate of 0.2%. They suggested that the sampling time is a critical factor in G6PD screening as samples drawn during or shortly after acute haemolysis will result in false-positive results. Another study was carried out by Goyal and group at the Division of Genetics of Maulana Azad Medical College and Lok Nayak Hospital, New Delhi, during a 2-year period from January 2011-December 2012. They screened 6,000 newborns for G6PD deficiency and reported an incidence of 1.5%.[5] Neonatal hyperbilirubinemia was present in 13.3% of cases. Of note, their study data showed 16% of the G6PD affected neonates to be females, which is a significant proportion. This further emphasizes the need for the implementation of a screening program for this condition. In another similar study carried out in Bangalore, a total of 41,027 babies were screened out of which 99 babies were confirmed to have G6PD deficiency.[15] In 15% of the G6PD deficient babies, neonatal hyperbilirubinemia was seen. Overall, their study reported a high incidence of 1 in 414 live births.
Sample Collection and Screening Tests for G6PD Deficiency
NBS studies carried out for G6PD in India have generally used heel-prick blood samples on filter paper strips. The recommended time of sampling is 24-48 hr after birth. There are a number of screening tests available for G6PD deficiency. The Beutler Fluorescent spot test is recommended by the International Committee for Standardization in Hematology for screening of G6PD deficiency.[35] The limitation of this test is the requirement of a UV light source which restricts its use in mass screening programs in resource-poor countries. There are two new screening methods recently developed that have a high sensitivity and specificity and also do not require a UV light source namely the color reduction test[36] and the modified Formazan ring test.[37] The latter test is suggested to be suitable for a screening program in India, as the test is rapid with results available in less than 24 hours, requires only basic laboratory equipment, and does not require extensive training of personnel. Also, it uses the same filter paper (Schleicher and Schuell #903C Duren, Germany) which is used in routine newborn screening for metabolic disorders thus allowing easy integration with screening programs for other inborn errors of metabolism.
Cost Benefit Analysis of NBS
Different countries have studied cost benefit analysis to implement NBS but such a study is not available in India. Nonetheless it is certain that NBS is not only meant to save lives but can also give societal economic benefit to the families, reduce National health burden, and give improved quality of life to the affected child and family. If we calculate the benefits of NBS, it is the net economic benefit from the program that is the monetary equivalent of avoided deaths and reductions of costs of care for complications due to delayed-diagnosis minus the additional costs of screening, diagnosis, and treatment associated with prompt diagnosis. Like Washington State Department of Health, Indian health system need to implement an approach for conducting evidence-based economic evaluations of above common disorders that can be included for the state-mandated newborn screening panel.
Conclusion
NBS in India has gained impetus in the last decade. There are several public and private NBS programs being carried out in different parts of the country. The key challenge is setting up a centralized policy to supervise the program in order to achieve maximum outcome from the same. Likewise, it is the need of the hour to create awareness among the medical community regarding this group of treatable conditions. Lastly, setting up the required infrastructure and employing trained personnel is critical for successful implementation of such a gross new-born screening program in every district of the country.
References:
1. Guthrie R. The origin of newborn screening. Screening. 1992;1(1):5-15. doi:10.1016/0925-6164(92)90025-Z
2. Lindner M, Gramer G, Haege G, Fang-Hoffmann J, Schwab KO, Tacke U, Trefz FK, Mengel E, Wendel U, Leichsenring M, Burgard P. Efficacy and outcome of expanded newborn screening for metabolic diseases – Report of 10 years from South-West Germany *. Orphanet J Rare Dis. 2011;6:44. doi:10.1186/1750-1172-6-44
3. Kapoor S, Thelma BK. Status of Newborn Screening and Inborn Errors of Metabolism in India. Indian J Pediatr. 2018;85(12):1110-1117. doi:10.1007/s12098-018-2681-5
4. ICMR Task Force on Inherited Metabolic Disorders. Newborn Screening for Congenital Hypothyroidism and Congenital Adrenal Hyperplasia. Indian J Pediatr. 2018;85(11):935-940. doi:10.1007/s12098-018-2645-9
5. Goyal M, Garg A, Goyal MB, Kumar S, Ramji S, Kapoor S. Newborn screening for G6PD deficiency: A 2-year data from North India. Indian Journal of Public Health. 2015;59(2):145. doi:10.4103/0019-557X.157537
6. Moinuddin M, Gagandeep V, Mutalik S. Prevalence of glucose 6 phosphate dehydrogenase (G6PD) deficiency in a community by newborn screening. International Journal of Contemporary Pediatrics. 2017;4:1018. doi:10.18203/2349-3291.ijcp20171719
7. Desai M, Vishwanath T, Sheth AR, Raikar RS. Neonatal screening for hypothyroidism–a review experience with thyrotropin estimation in the newborn. Indian Pediatr. 1978;15(6):463-467.
8. Rao NA, Devi ARR, Savithri HS, Rao SV, Bittles AH. Neonatal screening for amino acidaemias in Karnataka, South India. Clinical Genetics. 1988;34(1):60-63. doi:10.1111/ j.1399-0004.1988.tb02616.x
9. Ramadevi R, Savithri HS, Devi AR, Bittles AH, Rao NA. An unusual distribution of glucose-6-phosphate dehydrogenase deficiency of south Indian newborn population. Indian J Biochem Biophys. 1994;31(4):358-360.
10. Kaur G, Srivastav J, Jain S,Chawla D, Chavan BS, Atwal R, Randhawa G, Kaur A, Prasad R.. Preliminary report on neonatal screening for congenital hypothyroidism, congenital adrenal hyperplasia and glucose-6-phosphate dehydrogenase deficiency: a Chandigarh experience. Indian J Pediatr. 2010;77(9):969-973. doi:10.1007/s12098-010-0150-x
11. Sahai I, Zytkowicz T, Kotthuri SR, Kotthuri AL, Eaton RB, Akella RR. Neonatal Screening for Inborn Errors of Metabolism Using Tandem Mass Spectrometry: Experience of the Pilot Study in Andhra Pradesh, India. Indian J Pediatr. 2011;78(8):953-960. doi:10.1007/s12098-011-0398-9
12. Colaco MP, Desai MP, Ajgaonkar AR,Mahadik CV, VAS FE. Neonatal screening for hypothyroidism. Indian Pediatr. 1984;21(9):695-700.
13. Desai MP, Upadhye P, Colaco MP, Mehre M, Naik SP, Vaz FE, Nair N, Thomas M. Neonatal screening for congenital hypothyroidism using the filter paper thyroxine technique. Indian J Med Res. 1994;100:36-42.
14. Verma J, Roy P, Thomas DC, Jhingan G, Singh A, Bijarnia-Mahay S, Verma IC. Newborn Screening for Congenital Hypothyroidism, Congenital Adrenal Hyperplasia, and Glucose-6-Phosphate Dehydrogenase Deficiency for Improving Health Care in India. J Pediatr Intensive Care.2020;9(1):40-44. doi:10.1055/s-0039-1698424
15. Kommalur A, Devadas S, Kariyappa M, Sabapathy S, Benakappa A, Gagandeep V, Veranna Sajjan S, KrishnapuraLakshminarayana S, Dakshayani B, Devi Chinnappa G. Newborn Screening for Five Conditions in a Tertiary Care Government Hospital in Bengaluru, South India-Three Years Experience. J Trop Pediatr. 2020;66(3):284-289. doi:10.1093/tropej/fmz067
16. Fisher DA, Klein AH. Thyroid Development and Disorders of Thyroid Function in the Newborn. New England Journal of Medicine.1981;304(12):702-12.doi:10.1056/NEJM198103193041205
17. Walfish PG. Evaluation of three thyroid-function screening tests for detecting neonatal hypothyroidism. Lancet. 1976;1(7971):1208-1210. doi:10.1016/s0140-6736(76)92159-0
18. Delange F. Neonatal screening for congenital hypothyroidism: results and perspectives. Horm Res. 1997;48(2):51-61. doi:10.1159/000185485
19. Clayton PE, Miller WL, Oberfield SE, et al. Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the Lawson Wilkins Pediatric Endocrine Society. Horm Res. 2002;58(4):188-195. doi:10.1159/000065490
20. Belinda G, Vinay D, Moolechery J, Moolechery J, Mathew V, Anantharaman R, Ayyar V, Bantwal G. Congenital adrenal hyperplasia – experience from a tertiary centre in South India. Indian J Endocrinol Metab. 2012;16Suppl2:S385-S386. doi:10.4103/2230-8210.104102
21. Walia R, Singla M, Vaiphei K, Kumar S, Bhansali A. Disorders of sex development: a study of 194 cases. Endocr Connect. 2018;7(2):364-371. doi:10.1530/EC-18-0022
22. Devi ARR, Naushad SM. Newborn screening in India. Indian J Pediatr. 2004;71(2):157-160. doi:10.1007/BF02723099
23. Kishore Kumar R, Das H, Kini P. Newborn Screening for Congenital Adrenal Hyperplasia in India: What Do We Need to Watch Out for? J Obstet Gynaecol India. 2016;66(6):415-419. doi:10.1007/s13224-015-0712-y
24. Pearce M, Dauerer E, DiRienzo AG, Caggana M, Tavakoli NP. The influence of seasonality and manufacturer kit lot changes on 17α-hydroxyprogesterone measurements and referral rates of congenital adrenal hyperplasia in newborns. Eur J Pediatr. 2017;176(1):121-129. doi:10.1007/s00431-016-2814-7
25. González EC, Carvajal F, Frómeta A, Arteaga AL, Castells EM, Espinosa T, Coto R, Pérez PL, Tejeda Y, Del Río L, Segura MT. Newborn screening for congenital adrenal hyperplasia in Cuba: six years of experience. Clin Chim Acta.2013;421:73-78. doi:10.1016/j.cca.2013.02.020
26. Chennuri VS, Mithbawkar SM, Mokal RA, Desai MP. Serum 17 alpha hydroxyprogesterone in normal full term and preterm vs sick preterm and full term newborns in a tertiary hospital. Indian J Pediatr. 2013;80(1):21-25. doi:10.1007/s12098-012-0856-z
27. Vats P, Dabas A, Jain V, Seth A, Yadav S, Kabra M, Gupta N, Singh P, Sharma R, Kumar R, Polipalli SK. Newborn Screening and Diagnosis of Infants with Congenital Adrenal Hyperplasia. Indian Pediatr. 2020;57(1):49-55.
28. Speiser PW, Arlt W, Auchus RJ, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society* Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 2018;103(11):4043-4088. doi:10.1210/jc.2018-01865
29. Olgemöller B, Roscher AA, Liebl B, Fingerhut R. Screening for Congenital adrenal hyperplasia: adjustment of 17-hydroxyprogesterone cut-off values to both age and birth weight markedly improves the predictive value. J Clin Endocrinol Metab. 2003;88(12):5790-5794. doi:10.1210/jc.2002-021732
30. Frank JE. Diagnosis and management of G6PD deficiency. Am Fam Physician. 2005;72(7):1277-1282.
31. Mohanty D, Mukherjee MB, Colah RB. Glucose-6-phosphate dehydrogenase deficiency in India. Indian J Pediatr. 2004;71(6):525-529. doi:10.1007/BF02724295
32. Mezzacappa MA, Facchini FP, Pinto AC, et al. Clinical and genetic risk factors for moderate hyperbilirubinemia in Brazilian newborn infants. J Perinatol. 2010;30(12):819-826. doi:10.1038/jp.2010.48
33. Gómez-Manzo S, Marcial-Quino J, Vanoye-Carlo A, et al. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. Int J Mol Sci. 2016;17(12):2069. doi:10.3390/ijms17122069
34. Bubp J, Jen M, Matuszewski K. Caring for Glucose-6-Phosphate Dehydrogenase (G6PD)–Deficient Patients: Implications for Pharmacy. Pharmacy and Therapeutics. 2015;40(9):572.
35. Beutler E, Blume KG, Kaplan JC, Löhr GW, Ramot B, Valentine WN. International Committee for Standardization in Haematology: Recommended Screening Test for glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Br J Haematol. 1979;43(3):465-467. doi:10.1111/j.1365-2141.1979.tb03774.x
36. Kaplan M, Leiter C, Hammerman C, Rudensky B. Comparison of Commercial Screening Tests for Glucose-6-Phosphate Dehydrogenase Deficiency in the Neonatal Period. Clinical Chemistry. 1997;43(7):1236-1237. doi:10.1093/clinchem/43.7.1236
37. Padilla CD, Therrell BL. Newborn Screening in the Asia Pacific Region. J Inherit Metab Dis. 2007;30(4):490-506. doi:10.1007/s10545-007-0687-7
+


